
Case Report: Osteodysplastic Gerodermia in Panama
[Reporte de Caso: Gerodermia Osteodisplástica en Panamá]Indira Herrera Rodriguez1, Anyi Yu Pon1, Teresa Chávez Peña1, Mirna Chung1, Oleg Saldaña1
1. Hospital del Niño Dr. José Renán Esquivel, Department of Genetics, Panama, Rep. of Panama.
Downloads
Abstract
ABSTRACT: Gerodermia osteodysplastica (GO) is a rare genetic disease in which the inheritance is autosomal recessive within Cutis laxa disorders, it is characterized by a lax and wrinkled skin, osteoporosis leading to spontaneous fractures, congenital dislocation of the hips, hyperextensible joints, progeroid features, developmental delay and intellectual deficit. This is a condition caused by genetic mutations in the GORAB gene (1q24.2). We present the case of a 3-month-old infant with the typical phenotype of this condition, lax and wrinkled skin, specially the hands and feet, in addition to a knee dislocation. A panel testing for connective tissue disorders was performed, which identified an alteration in the GORAB gene, confirming the diagnosis de Gerodermia osteodysplastica.
INTRODUCTION
Osteodysplastic geroderma is an autosomal recessive genetic disorder caused by a mutation in the GORAB gene (1q24.2), it is found within the Cutis laxa disorders, in recent studies mutations have been identified in patients with similar clinical phenotypes in the PYCR1 gene (17q25.3).
This disorder was first described by Bamatter et al. in 1950 in members of a family of Swiss origin, at the time they saw similarities between them and Walt Disney characters and so they called it Walt Disney dwarfism [4]. Later studies were also based on this family and it was concluded that the disorder was hereditary, in addition to emphasizing the characteristics and clinical manifestations included in this disorder.
The phenotype of this disorder is very similar to other connective tissue pathologies, among which are the cutis laxa syndromes.
Caso Clínico: Gerodermia Osteodisplástica
Infant under 3 months of age, female, first child of a 15-year-old adolescent mother, who had 2 prenatal check-ups, no infections or complications during pregnancy, parents not related by blood according to the mother, family history denied. The patient was born at 40 weeks of gestation, via vaginal delivery without complications. APGAR 9/9, birth weight 2.6 kg. During her initial evaluation they noted a lax, loose and redundant skin, giving the impression of an aged skin appearance, in addition to long fingers. The patient was discharged with her mother.
She was referred to the Genetics outpatient clinic since her neonatal period due to the findings on physical examination with suspicion of a connective tissue disease. In her first evaluation the typical phenotype of this syndrome was found, an elongated fascia, with a lax, dry skin, hyperlaxity, limbs with arachnodactyly, in addition she had right knee subluxation for which she was sent for evaluation with Orthopedics.
Initially, peripheral blood karyotype was performed with a result of 46 XX normal female. A molecular study with a genetic panel of connective tissue disorders was requested and confirmed the diagnostic suspicion of osteodysplastic geroderma, with mutation in the GORAB gene, variant c.743G>C (p.Arg248Pro) in homozygote.
Examen físico actual.
Weight of 4.69 kg, size of 57 cm and head circumference of 37.5 cm.
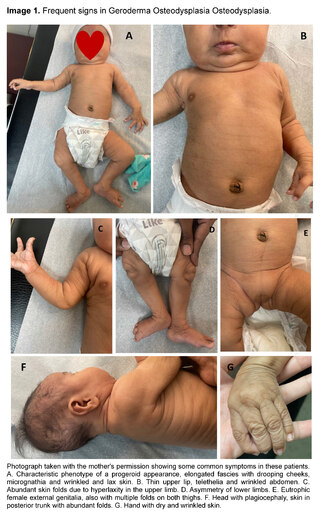
Alert, active and reactive to stimuli. Progeroid appearance. He had adequate cephalic support. Head with plagiocephaly, we palpate normotensive anterior fontanel, elongated fascies, a lax, dry and wrinkled skin, especially at the level of hands, feet and back, in addition to sparsely populated eyebrows, eyes with symmetrical pupils and preserved extraocular movements, follows with the gaze, low-set and large pinnae, bulbous nose, with anteverted nostrils, elongated philtrum, small mouth, with thin upper lip, high palate, micrognathia, neck with redundant skin, telethelia, abdomen with wrinkled appearance, soft and depressible, without palpable visceromegaly, female external genitalia apparently eutrophic, extremities with long fingers, hyperlaxity of joints, asymmetrical lower limbs, impressing left leg longer than right. Distal neurovascular was preserved.
Up to this moment with an adequate psychomotor development for her age.
We present some images of our patient, all with prior authorization of the mother for academic reasons.
Evaluation by the Cardiology service:
- Normal evaluation and discharged.
Evaluation by the Orthopedics service:
- He remains on multidisciplinary follow-up with physical therapy and rehabilitation, early stimulation and orthopedics.
DISCUSSION
Osteodysplastic geroderma is a rare autosomal recessive connective tissue pathology of genetic origin with an incidence of < 1/1,000,000 and is more frequent in the Middle Eastern population. It was initially described by Bamatter et al. in a Swiss family and later more publications of this same family appeared. Knowing that it is an entity due to a genetic mutation, it is important to approach families and offer them timely genetic counseling.
Multiple cases have been reported in Panama. So far, 2 cases have been reported at the Hospital del Niño Dr. José Renán Esquivel.
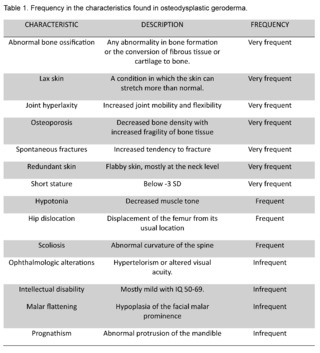
The clinical presentation is quite variable, it can be seen from birth, it is characterized by the presence of facial dysmorphism, aged appearance of the skin, which is thin, hyperlax, with abundant folds and redundant, abnormal bone ossification, osteoporosis, recurrent fractures, hip dislocation, hyperlaxity of joints, short stature, growth retardation, hypotonia, ocular and visual disturbances, sometimes there are mild degrees of intellectual disability. Table 1 shows the frequency of presentation of each one [3].
In cases of severe osteopenia, where recurrent spontaneous fractures occur, the use of bisphosphonates has been recommended to help reduce these episodes. The management of this pathology requires a multidisciplinary approach with Orthopedics, Ophthalmology, Dermatology, Physical Therapy and Rehabilitation, Early Stimulation, Genetics and Mental Health.
These patients have a good prognosis and a normal life expectancy in most cases. As the person grows, there is improvement of symptoms and decrease of fracture episodes since there is strengthening of the bones and fewer falls or accidents occur.
References
[1] Orphanet: Gerodermia osteodisplástica. INSERM US14. Available from: https://www.orpha.net/consor4.01/www/cgi-bin/OC_Exp.php?lng=ES&Expert=2078
[2] Hennies HC, Kornak U, Zhang H, Egerer J, Zhang X, Seifert W, et al. Gerodermia osteodysplastica is caused by mutations in SCYL1BP1, a Rab-6 interacting golgin. Nature Genetics. 2008 Nov 9;40(12):1410-2. DOI: https://doi.org/10.1038/ng.252
[3] Geroderma osteodysplastica - About the Disease - Genetic and Rare Diseases Information Center. rarediseases.info.nih.gov. [cited 2024 Jan 20]. Available from: https://rarediseases.info.nih.gov/diseases/413/geroderma-osteodysplastica
[4] OMIM Database, Entry - #231070 - GERODERMA OSTEODYSPLASTICUM; Available from: https://www.omim.org/entry/231070
[5] ClinVar Miner Database, List of variants reported ad benign for Geroderma osteodysplastica. Available from: https://clinvarminer.genetics.utah.edu/variants-by-condition/Geroderma%20osteodysplastica/significance/benign
[6] Alotaibi M., Aldhubaiban D., Alasmari A., Alotaibi L. A Case of Geroderma Osteodysplasticum Syndrome: Unique Clinical Findings. Journal of Clinical Research and Reports. 2021 Oct. Available from: https://www.auctoresonline.org/uploads/articles/1637307023JCRR-21-CR-207_Galley_Proof.pdf. DOI: https://doi.org/10.31579/2690-1919/207
[7] Bamatter F, Franceschetti A,Klein D, Sierro A: Gerodermie osteodysplastique hereditaire. AnnPediat 1950; 174: 126-127
[8] R. Glorio, A. Solari, R. Haas, S. Carbia, B. Olivieri, S. D. Gregorio, A. Woscoff: Gerodermia osteodisplásica. Revista de Pediatría Argentina. Vol VIII - No. 3 - 2002.
[9] Human Phenotype Ontology, Accessed Feb 2024. Available from: https://hpo.jax.org/app/
